Software
-

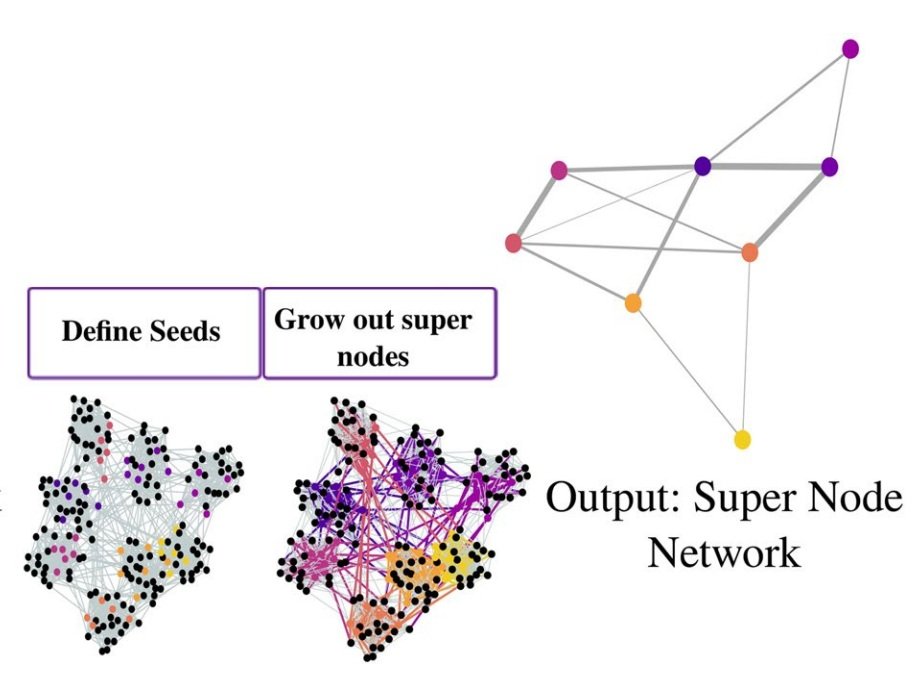
ALS peripheral immune diagnostic
We developed a diagnostic model from immune cells in peripheral blood, which can stratify slow and fast progressing ALS patients. The model was trained in a cohort profiled with CyTOF and generalizes to an independent cohort profiled with scRNA-seq. We identified dysregulated immune coordination patterns in fast progressing ALS donors. Work led by Luvna Dhawka and Baggio Evangelista.
Preprint ; Code ; Processed Data (annData for CyTOF and scRNA-seq)
-
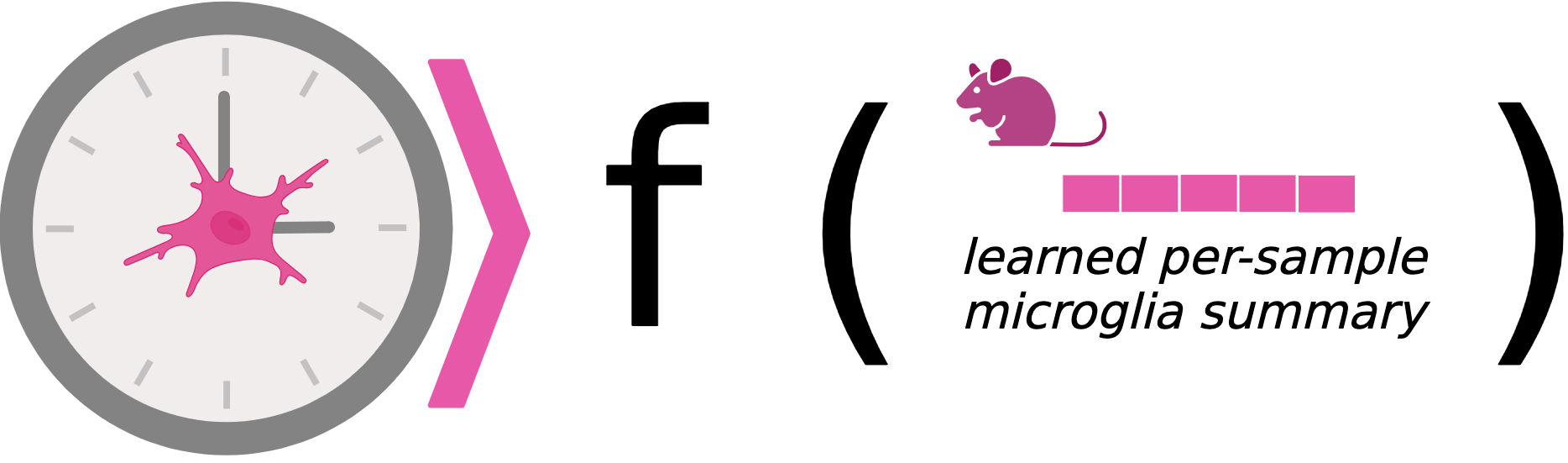
Microglia aging clock
Microglia are key innate immune cells in the brain and have shown to driving aging and neurodegeneration. We leveraged single-cell datasets to create robust microglia aging clocks. Our results show that there are characteristic changes in genetic programs that drive aging trajectories. This work is in collaboration with Anthony Zannas.
(Aging Cell, 2025)
-
-
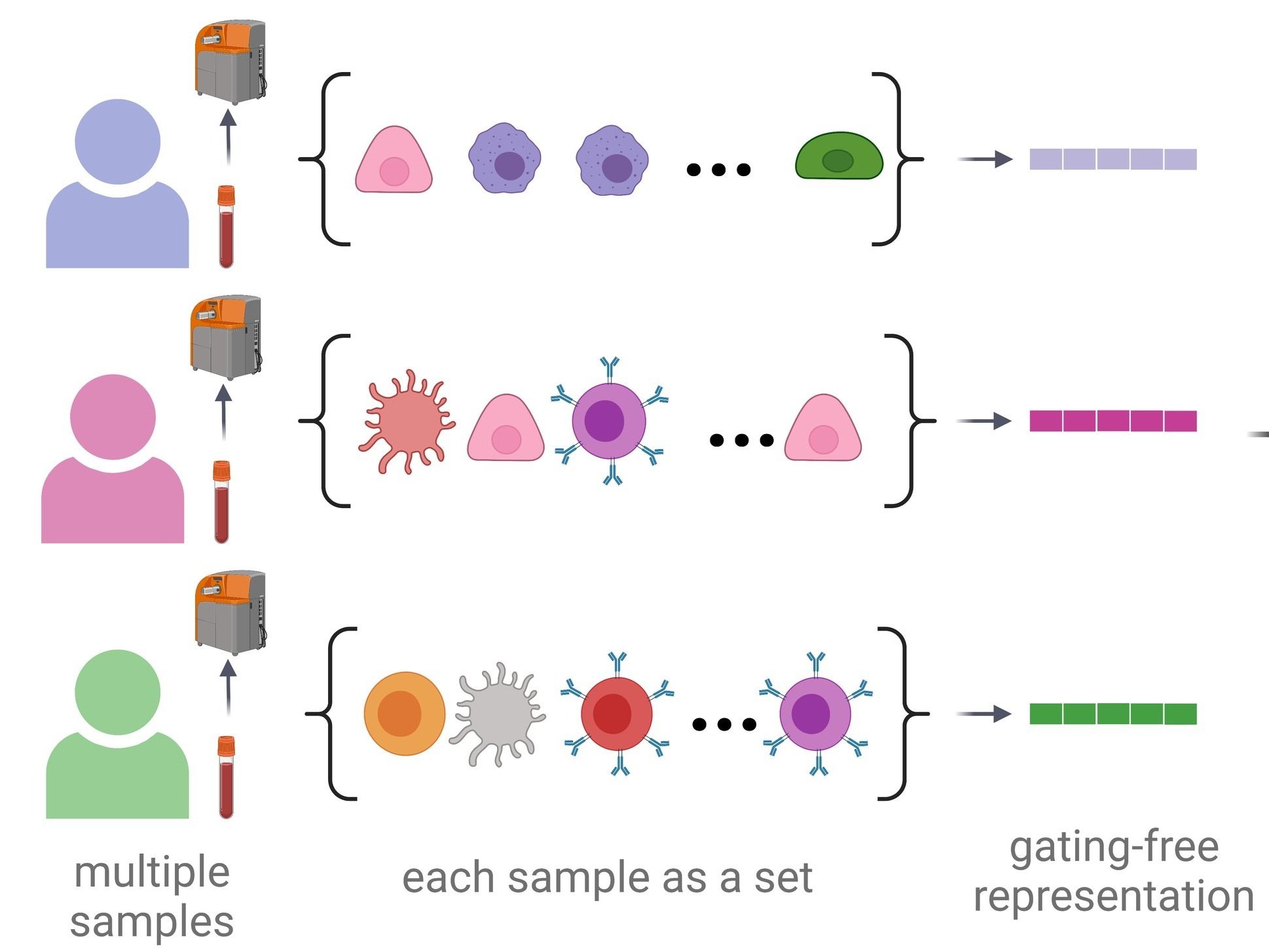
CytoSet + CytoCoSet
CytoSet and CytoCoSet are two variants of our original CytoSet approach to learn gating-free representations of single-cell data for prediction or outcome or phenotype. CytoCoSet incorporates extra covariates that are independent of the single-cell information. Works led by Haidong Yi (CytoSet, ACM BCB, 2021) and Chi-Jane Chen (CytoCoSet, BMC Bioinformatics, 2025).
-
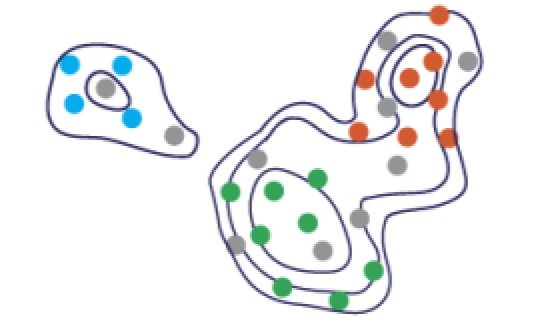
Single-Cell Sketching
‘Sketching’ means good downsampling for single-cell data. In immunology, and especially for working with flow and mass cytometry data, we want to preserve all major cell-types and their relative frequencies. Our sketches based on Kernel Herding do this very effectively! Work led my Vishal Baskaran and Jolene Ranek in collaboration with Junier Oliva (ACM BCB 2022).
-
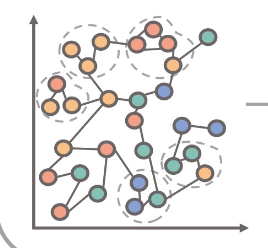
Cytocoarsening
Many single-cell analysis algorithms for linking immune cells to clinical outcomes rely on a graph representation of the data. To make such analyses more scalable, we developed a single-cell graph coarsening approach where redundant cells are aggregated based on graph structure and clinical outcomes. Work led by Chi-Jane Chen. (PSB,2023)
-
-
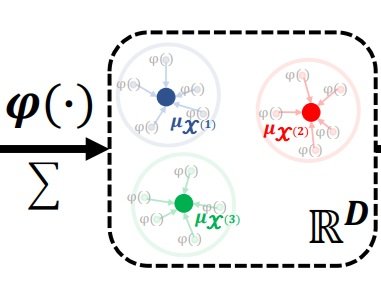
CKME
Finding prototypical cells for a clinical phenotype using kernel mean embedding. In addition, we use a random Fourier feature representation for each cell and a mean embedding approach to define a single vector representation for each profiled sample. Work led by Siyuan Shan and in collaboration with Junier Oliva (Under Review, 2022).
-
-